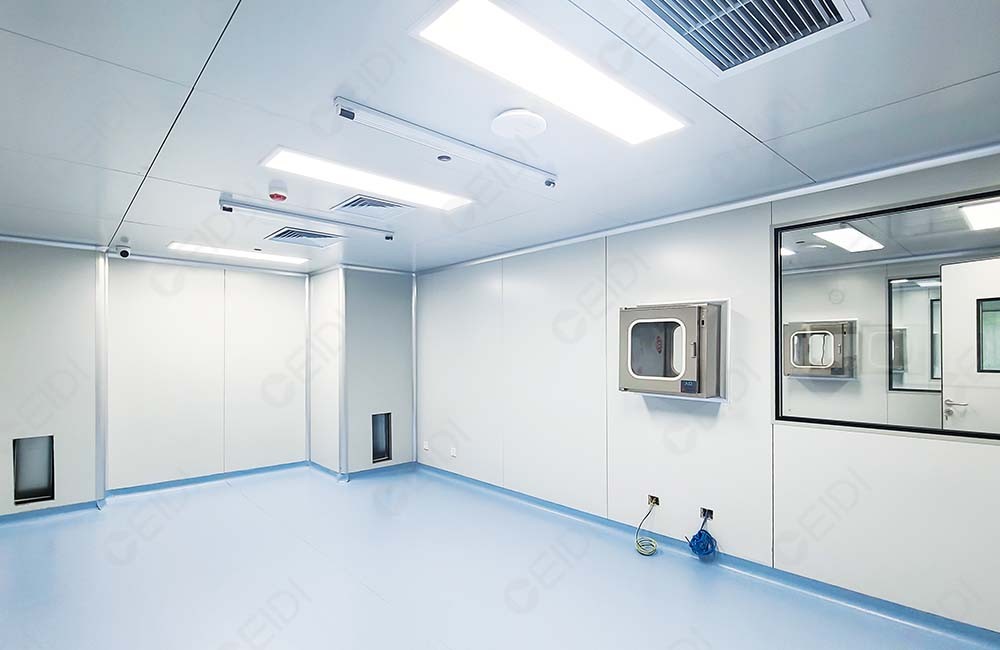
净化工程
精密制造净化工程
推荐设备




体外诊断试剂作为疾病筛查、诊断和治疗监测的重要工具,其生产质量直接关系到临床检测的准确性和患者安全。近年来,随着国家药监局《医疗器械生产质量管理规范附录体外诊断试剂》等法规的持续更新,以及行业对产品溯源性和稳定性的高标准要求,建设符合GMP标准的洁净车间已成为体外诊断企业的刚需。体外诊断试剂到中试及生产环节,就需要在GMP洁净厂房的条件下进行。体外诊断试剂应符合《体外诊断试剂生产实施细则(试行)》的要求...
体外诊断试剂作为疾病筛查、诊断和治疗监测的重要工具,其生产质量直接关系到临床检测的准确性和患者安全。近年来,随着国家药监局《医疗器械生产质量管理规范附录体外诊断试剂》等法规的持续更新,以及行业对产品溯源性和稳定性的高标准要求,建设符合GMP标准的洁净车间已成为体外诊断企业的刚需。体外诊断试剂到中试及生产环节,就需要在GMP洁净厂房的条件下进行。体外诊断试剂应符合《体外诊断试剂生产实施细则(试行)》的要求。平面布局要求合理紧凑,人流、物流路线清晰明了;参观、消防通道畅通。每个净化级别符合生产需求;洁净区、人员净化、物料净化和其他辅助用房分区布置,同时考虑生产操作、工艺设备安装和维修、管线布置、气流以及净化空调系统各种技术设施的综合协调;不同空气洁净度等级房间之间联系频繁时,设有防止污染的措施,如缓冲间、气闸室、传递窗等。

CEIDI西递设计组会与使用方技术负责人明确生产工段所处的环境要求,对有空气净化级别的工序进行净化设计,背景净化环境按照以下要求设置:
1.普通化学类诊断试剂的生产可以在清洁环境中进行。
2.阴性、阳性血清、质粒或血液制品的处理操作可以设计C级环境,与相邻区域保持相对负压,并符合防护规定。
3.酶联免疫吸附试验试剂、免疫荧光试剂、免疫发光试剂、聚合酶链反应(PCR)试剂、金标试剂、干化学法试剂、细胞培养基、校准品与质控品、酶类、抗原、抗体和其他活性类组分的配制及分装等产品的配液、包被、分装、点膜、干燥、切割、贴膜,以及内包装等工艺环节,至少控制于D级净化环境 。
4.无菌物料的分装工艺段环境设计为局部B级。

在实际项目推进中,环境建设的达成是由多个技术系统共同组成,其中包括但不限定于装饰部分的围合施工、空调及排风系统的净化配置、压差系统的控制等等。每个项目的具体使用端要求的不同,就如航行的大船需要舵手一般,需要有经验的和资质的设计单位与施工单位合作进行。当然如果将设计、施工甚至采购都能三位一体的统筹总包,之于精准医疗行业这类对环境有专业要求的场所来说,是较为理想的建设模式。
只需使用方给出明确的具有污染性、传染性和高生物活性的物料,明确生产过程中会有哪些风险,CEIDI西递设计组就可以此为依据制定合适的技术方案及空间环境受控方式,有效避免造成传染、污染或泄漏等。几个特殊的类别需要特别建设受控环境,列举如下:
1.高风险的生物活性物料的操作我们会设计分割单独用室,且使用单独的空气净化系统,与相邻区域保持负压,排出的空气不进循环系统,通过专业净化处理后按指定路径排出。
2.进行危险度二级及以上的病原体操作用室,CEIDI西递会予以配置恰当的生物安全柜,将空气进行有效处理后排出。
3.特殊的高致病性病原体的采集、制备,根据标准规范按照卫生部颁布的行业标准《微生物和生物医学实验室生物安全通用准则》等相关规定,装备全套P3级实验室相应设施。
4. 激素类试剂组分的洁净室(区)我们推荐采用独立的专用的空气净化系统,排出的空气不进循环系统,通过专业净化处理后按指定路径排出。
5. 强毒微生物操作区、芽孢菌制品操作区应与相邻区域保持相对负压,配备独立的空气净化系统。
6.生产聚合酶链反应(PCR)试剂的,其生产和检验应当在独立的建筑物或空间内进行,受控环境建设要保证空气不直接联通,防止扩增时形成的气溶胶造成交叉污染。

当然,也有体外诊断试剂品类对生产环境没有空气净化要求的,环境设置为清洁环境即可,在此环境内完成工艺流程。除了以上几个核心受控环境需要做全方位的技术方案外,还要重视原料或者参考品、物料的存储环境。譬如具有传染性或来源于生物体的物料如含阳性病原体的参考品、动物腹水或血清、人体样品(体液、血液等)等物料,要按照 中华人民共和国国务院令第424号《病原微生物实验室生物安全管理条例》,卫生部《人间传染的病原微生物名录》等要求,设计符合具体产品储存要求和防护要求的空间环境。CEIDI西递在医学诊断实验及洁净车间工程案例较多,有需要的企业可以联系我们获取详细方案及报价。



相关阅读:

全国服务热线
地址:上海市虹桥商务区富力环球中心